Table of Contents
Genome Annotation
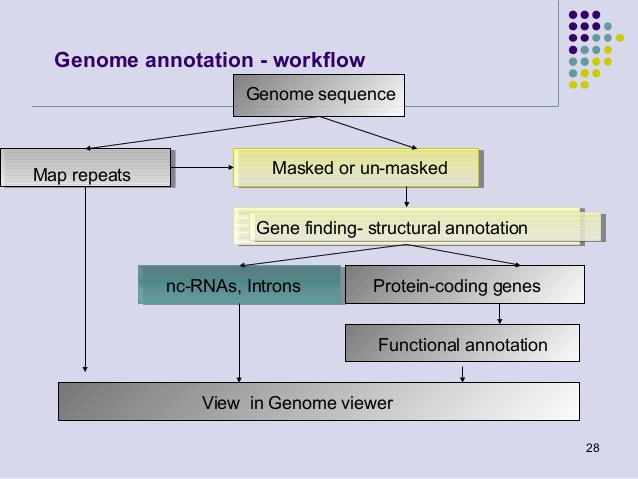
Genome annotation is a key process for identifying the coding and non-coding regions of a genome, gene locations and functions. Analysis of DNA sequence with genome annotation software tools allow finding and mapping genes, exons-introns, regulatory elements, repeats and mutations. Genome databases are essential to retrieve information on gene name, protein product and DNA sequence functions.
Gene Ontology Annotation
BLASTX
The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families.A BLAST search enables a researcher to compare a query sequence with a library or database of sequences, and identify library sequences that resemble the query sequence above a certain threshold.
Different types of BLASTs are available according to the query sequences. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence. The BLAST algorithm and program were designed by Stephen Altschul, Warren Gish, Webb Miller, Eugene Myers, and David J. Lipman at the National Institutes of Health and was published in the Journal of Molecular Biology in 1990 and cited over 50,000 times.
 Input: Input sequences (in FASTA or Genbank format) and weight matrix.
Input: Input sequences (in FASTA or Genbank format) and weight matrix.
Output: BLAST output can be delivered in a variety of formats. These formats include HTML, plain text, and XML formatting.
The details about process and algorithm coule be found here and official website
NCBI BLAST:
BLAST stands for Basic Local Alignment Search Tool.The emphasis of this tool is to find regions of sequence similarity, which will yield functional and evolutionary clues about the structure and function of your sequence.
This tool can be used in the following contexts:Protein, Nucleotide, Vectors.
PSI-BLAST:
Position specific iterative BLAST (PSI-BLAST) refers to a feature of BLAST 2.0 in which a profile is automatically constructed from the first set of BLAST alignments. PSI-BLAST is similar to NCBI BLAST2 except that it uses position-specific scoring matrices derived during the search, this tool is used to detect distant evolutionary relationships. PHI-BLAST functionality is available to use patterns to restrict search results.
Publications
- (Altschul et al., 1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. PMID: 9254694
- (Altschul et al., 1990) Basic local alignment search tool. J Mol Biol. PMID: 2231712
Institution(s)
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, USA
citation: Sequence Similarity Searching
Blast2GO
Permits functional annotation, management, and data mining of novel sequence data. Blast2GO is based on the utilization of common controlled vocabulary schemas, the gene ontology (GO). It takes in consideration similarity, the extension of the homology, the database of choice, the GO hierarchy, and the quality of the original annotations. This tool is suitable for plant genomics research. It generates functional annotation and assesses the functional meaning of their experimental results.

Publications:
- (Conesa et al., 2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research.
PMID: 16081474 DOI: 10.1093/bioinformatics/bti610 - (Conesa and Götz, 2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics.
PMID: 18483572 DOI: 10.1155/2008/619832 - (Götz et al., 2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res.
PMID: 18445632 DOI: 10.1093/nar/gkn176 - (Aparicio et al., 2006) Blast2GO goes grid: developing a grid-enabled prototype for functional genomics analysis. Stud Health Technol Inform.
PMID: 16823138
Institutions(s): Bioinformatics Department, Centro de Investigación Príncipe Felipe, Valencia, Spain
GO Semantic Similarity Analysis
G-SESAME | Gene Semantic Similarity Analysis and Measurement Tools
A set of online tools for measuring the semantic similarities of Gene Ontology (GO) terms and the functional similarities of gene products, and for further discovering biomedical knowledge from the GO database. Visualization techniques are provided in these tools to allow users to inspect the locations of the GO terms within the GO graph and to visually determine the semantic similarity. A batch command interface is also provided for users to execute the tools to measure the semantic similarity of a group of GO terms or functional similarities of a group of genes. Web based APIs are also provided for advanced users.
Publications:
- (Song, 2014) Measure the Semantic Similarity of GO Terms Using Aggregate Information Content. IEEE Trans Comput Biol Bioinform.
PMID: 26356015 DOI: 10.1109/TCBB.2013.176 - (Du et al., 2009) G-SESAME: web tools for GO-term-based gene similarity analysis and knowledge discovery. Nucleic Acids Res.
PMID: 19491312 DOI: 10.1093/nar/gkp463 - (Wang et al., 2007) A new method to measure the semantic similarity of GO terms. Bioinformatics.
PMID: 17344234
Institutions(s): School of Computing, Clemson University, Clemson, SC, USA
clusterProfiler | GO semantic similarity analysis : Genome annotation
Automates the process of biological-term classification and the enrichment analysis of gene clusters. ClusterProfiler supports three species, including humans, mice, and yeast. It offers a gene classification method, namely groupGO, to sort genes based on their projection at a specific level of the gene ontology (GO) corpus. This tool is able to calculate enrichment test for GO terms and KEGG pathways based on hypergeometric distribution.
Publications:
- (Yu et al., 2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS.
- (Yu, 2018) clusterProfiler: An universal enrichment tool for functional and comparative study. BioRxiv.
Institutions(s): State Key Laboratory of Emerging Infectious Diseases and Centre of Influenza Research, School of Public Health, The University of Hong Kong, Hong Kong, China
GOToolBox | GO semantic similarity analysis : DNA sequence analysis
Provides a series of programs allowing the functional investigation of groups of genes, based on the Gene Ontology resource. GOToolBox allows 1) the identification of statistically relevant over- or under-represented terms in a gene dataset, 2) the clustering of functionally related genes within a set and 3) the retrieval of genes sharing annotations with a query gene. The user can also constrain the GO annotations to a slim hierarchy or to a given level of the ontology, in order to facilitate the interpretation of the results.
Publications:
- (Martin et al., 2004) GOToolBox: functional analysis of gene datasets based on Gene Ontology. Genome Biol.
Institutions(s): Laboratoire de Génétique et Physiologie du Développement, IBDM, CNRS/INSERM/Université de la Méditerranée, Parc Scientifique de Luminy, Marseille, France
SML-Toolkit | Semantic Measures Library Toolkit
A Java Toolkit dedicated to semantic measures computation and analysis. SML-Toolkit is composed of various tools related to semantic measures computation and analysis. Those tools are provided through a common command-line interface.
Publications:
- (Harispe et al., 2014) A framework for unifying ontology-based semantic similarity measures: a study in the biomedical domain. J Biomed Inform.
- (Harispe et al., 2014) The semantic measures library and toolkit: fast computation of semantic similarity and relatedness using biomedical ontologies. Bioinformatics.
Institutions(s): LGI2P/EMA Research Centre, Site EERIE, Parc Scientifique G Besse, Nîmes, France
Promoter Predication
FirstEF | First Exon Finder
A 5’ terminal exon and promoter prediction program. FirstEF consists of different discriminant functions structured as a decision tree. The probabilistic models are optimized to find potential first donor sites and CpG-related and non-CpG-related promoter regions based on discriminant analysis. For every potential first donor site (GT) and an upstream promoter region, FirstEF decides whether or not the intermediate region can be a potential first exon, based on a set of quadratic discriminant functions.
Publications:
- (Davuluri et al., 2001) Computational identification of promoters and first exons in the human genome. Nat Genet.
Institutions(s): Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, NY, USA
CpGpromoter
A program for a large-scale human promoter mapping using CpG islands. CpGpromoter is based on results of discriminant analysis between the promoter-associated CpG islands and non-associated ones. It enables an efficient mapping of human promoters with 2Kb resolution, if there is a CpG island inside an interval (-500…+1,500) around a transcription start site.
Publications:
- (Ioshikhes and Zhang, 2000) Large-scale human promoter mapping using CpG islands. Nat Genet.
Institutions(s): Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA
PROmiRNA
A program for annotating miRNA promoters in human, as well as other species. PROmiRNA uses deepCAGE data from the FANTOM4 Consortium and integrated cage tag counts and other promoter features, such as CpG content, conservation and TATA box affinity, to score the potential of a candidate region to be a promoter. Given a list of genomic regions of interest, in the form of a gff file, PROmiRNA returns the most probable promoter locations, together with the posterior probabilities calculated by the model.
Publications:
- (Marsico et al., 2013) PROmiRNA: a new miRNA promoter recognition method uncovers the complex regulation of intronic miRNAs. Genome Biol.
Institutions(s): Max Planck Institute for Molecular Genetics, Berlin, Germany; Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
Annotation Workflows
RAST | Rapid Annotation using Subsystem Technology
Assists in annotating complete or nearly complete bacterial and archaeal genomes. RAST is a fully-automated application provides high quality genome annotations for these genomes across the whole phylogenetic tree. It includes a user interface that allows registered users to make manual changes to their genomes before retrieving them. It was designed to extend annotations to as many protein-encoding genes in as many genomes as possible.
Publications:
- (Overbeek et al., 2014) The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res.
- (Aziz et al., 2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics.
Prokka | Annotation workflows
A command line software tool to fully annotate a draft bacterial genome in about 10 min on a typical desktop computer. It produces standards-compliant output files for further analysis or viewing in genome browsers. Prokka uses parallel processing to decrease running time on multicore computers. The most time-consuming steps are BLAST+ and hmmscan, which both support multiple CPUs natively. However, Prokka is more efficient if it runs multiple single CPU threads on subsets of the data, which it achieves using GNU parallel.
Publications:
- (Seemann, 2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics.
Institutions(s): Victorian Bioinformatics Consortium, Monash University, Clayton; Life Sciences Computation Centre, Victorian Life Sciences Computation Initiative, Carlton, Australia
BlastKOALA
Assigns K numbers to the user’s sequence data by BLAST searches, respectively, against a nonredundant set of KEGG GENES. KOALA (KEGG Orthology And Links Annotation) is KEGG’s internal annotation tool for K number assignment of KEGG GENES using SSEARCH computation. Annotate Sequence in KEGG Mapper and Pathogen Checker in KEGG Pathogen are special interfaces to this server and can be executed in an interactive mode. BlastKOALA is suitable for annotating fully sequenced genomes.
Publications:
- (Kanehisa et al., 2016) BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J Mol Biol.
Institutions(s): Institute for Chemical Research, Kyoto University, Uji, Kyoto, Japan; Healthcare Solutions Department, Fujitsu Kyushu Systems Ltd, Hakata-ku, Fukuoka, Japan; Institute for Chemical Research, Kyoto University, Uji, Kyoto, Japan