Table of Contents
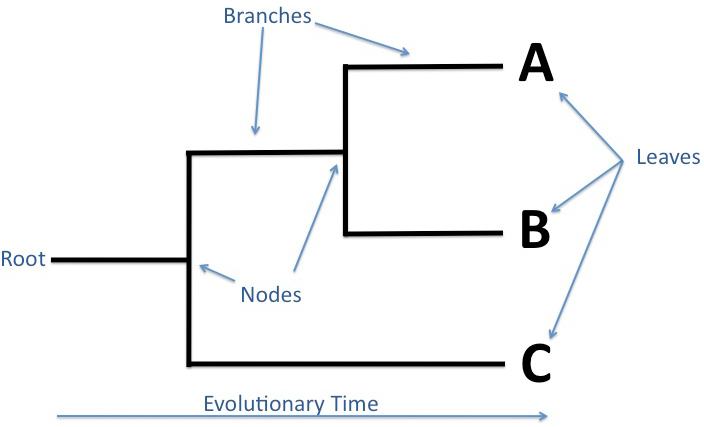
In biology, phylogenetics is the study of the evolutionary history and relationships among individuals or groups of organisms (e.g. species, or populations). These relationships are discovered through phylogenetic inference methods that evaluate observed heritable traits, such as DNA sequences or morphology under a model of evolution of these traits. The result of these analyses is a phylogeny (also known as a phylogenetic tree) – a diagrammatic hypothesis about the history of the evolutionary relationships of a group of organisms.
Phylogenies are important for addressing various biological questions such as relationships among species or genes, the origin and spread of viral infection and the demographic changes and migration patterns of species. The advancement of sequencing technologies has taken phylogenetic analysis to a new height. Phylogenies have permeated nearly every branch of biology, and the plethora of phylogenetic methods and software packages that are now available may seem daunting to an experimental biologist.
Phylogenetic inference
The task of resolving the tree of life of extant species remains one of the grand challenges in evolutionary biology. As the number of trees grows superexponentially with the number of species for which an evolutionary tree is reconstructed, tree inference is considered a hard problem in computer science. The plethora of algorithmic challenges associated with phylogenetic trees and their efficient computation gave rise to the discipline of “phyloinformatics.”
MEGA
An integrated tool for conducting sequence alignment, inferring phylogenetic trees, estimating divergence times, mining online databases, estimating rates of molecular evolution, inferring ancestral sequences, and testing evolutionary hypotheses.
Publications
- (Kumar et al., 1994) MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput Appl Biosci.
- (Kumar et al., 2001)MEGA2: molecular evolutionary genetics analysis software. Bioinformatics.
- (Tamura et al., 2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol.
- (Kumar et al., 2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol.
Institution(s)
Institute for Genomics and Evolutionary Medicine, Temple University Department of Biology, Temple University Center for Excellence in Genome Medicine and Research, King Abdulaziz University, Jeddah, Saudi Arabia.
ADAPTML
Allows identification of populations as groups of related strains sharing a common projected habitat, which reflects their relative abundance in the measured environmental categories. ADAPTML maps changes in environmental preference onto the tree by predicting projected habitats for each extant and ancestral strain in the phylogeny. It builds a hidden Markov model for the evolution of habitat associations.
Publications
- (2008 Science) esource partitioning and sympatric differentiation among closely related bacterioplankton.
Institution(s)
Department of Civil and Environmental Engineering, MA USA; Institute of Technology (MIT), Cambridge, MA, USA; Computational and Systems Biology Initiative, MIT, Cambridge, MA, USA
Phylogenetic Network Construction
The evolutionary history of a set of species is usually described by a rooted phylogenetic tree. Although it is generally undisputed that bifurcating speciation events and descent with modifications are major forces of evolution, there is a growing belief that reticulate events also have a role to play. Phylogenetic networks provide an alternative to phylogenetic trees and may be more suitable for data sets where evolution involves significant amounts of reticulate events, such as hybridization, horizontal gene transfer, or recombination.
CLC bio
Allows to analyze, compare, and visualize next generation sequencing (NGS) data. CLC Genomics Workbench offers a complete and customizable solution for genomics, transcriptomics, epigenomics, and metagenomics. The software enables to generate custom workflows, which can combine quality control steps, adapter trimming, read mapping, variant detection, and multiple filtering and annotation steps into a pipeline.
Citation Publications
- (2018 PMCID: 5959919) Phenotypic diversification by enhanced genome restructuring after induction of multiple DNA double-strand breaks
Documentation
SPRDist
Solves the rooted Subtree Prune and Regraft (rSPR) distance problem for many datasets. SPRDist is a method that uses integer programming to resolve the problem of computing rSPR distances between two rooted binary trees. This application takes two binary phylogenetic trees and computes the smallest number of (SPR) operations that transform one tree to the other. It only uses a publicly available integer linear programming (ILP) solver.
Publications
- (2009 Bioinformatics) A practical method for exact computation of subtree prune and regraft distance.
Documentation
Institution(s)
Department of Computer Science and Engineering, University of Connecticut, Storrs, CT, USA
Discrete Character Evolution
The study of discrete characters is crucial for the understanding of evolutionary processes. Even though great advances have been made in the analysis of nucleotide sequences, computer programs for non-DNA discrete characters are often dedicated to specific analyses and lack flexibility. Discrete characters often have different transition rate matrices, variable rates among sites and sometimes contain unobservable states.
CAFE
Enables the accurate estimation of rates of gene family evolution when there are errors in the observed gene family sizes. By allowing users to marginalize over the uncertainty in the observed gene family sizes, CAFE 3 provides a platform for expanding comparative genomic analyses into clades consisting solely of draft genome sequences.
Publications
- (2013 Mol Biol Evol) Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3.
Documentation
Institution(s)
K.U.Leuven, OKP Research GroupTiensestraat, Leuven, Belgium; Department of Statistics, U.C. Davis Kerr Hall, Davis, CA, USA
DupliPHY
A software tool to determine the evolutionary histories of gene families over a phylogenetic tree. Given a set of gene family sizes and a phylogenetic tree DupliPHY will calculate the ancestral family sizes at each internal node within the tree. DupliPHY returns a list of ancestral family sizes and a phylogenetic tree for each family with the ancestral family sizes listed as internal node labels.
Publications
- (2015 Bioinformatics) DupliPHY-Web: a web server for DupliPHY and DupliPHY-ML
Institution(s)
Faculty of Life Sciences, University of Manchester, Oxford Road, Manchester, UK
Tumor Progression
Cancer can be a result of accumulation of different types of genetic mutations such as copy number aberrations. The data from tumors are cross-sectional and do not contain the temporal order of the genetic events. Finding the order in which the genetic events have occurred and progression pathways are of vital importance in understanding the disease.
CT-CBN
A specific probabilistic graphical model for the accumulation of mutations and their interdependencies. The Bayesian network models cancer progression by an explicit unobservable accumulation process in time that is separated from the observable but error-prone detection of mutations. Model parameters are estimated by an expectation-maximization algorithm and the underlying interaction graph is obtained by a simulated annealing procedure.
Publications
(2011 PLoS One) The temporal order of genetic and pathway alterations in tumorigenesis.
(2009 Bioinformatics) Quantifying cancer progression with conjunctive Bayesian networks
Institution(s)
Department of Biosystems Science and Engineering, ETH Zurich, Basel, Switzerland; Department of Mathematics, North Carolina State University, Raleigh, NC, USA
TRONCO
Infers cancer progression models from heterogeneous genomic data. TRONCO is an R package built to extract population-level models describing the trends of accumulation of alterations in a cohort of cross-sectional samples and individual-level models that reveal the clonal evolutionary history in single cancer patients, when multiple samples are available. It also implements an oncoprint system to visualize the processed data.
Publications
- (2017 BioRxiv) Learning mutational graphs of individual tumor evolution from multi-sample sequencing data.
Institution(s)
Department of Pathology, Stanford University, CA, USA; Department of Informatics, Systems and Communication, University of Milan-Bicocca, Milan, Italy
Tree Visualization
Phylogenetic trees are pervasively used to depict evolutionary relationships. Increasingly, researchers need to visualize large trees and compare multiple large trees inferred for the same set of taxa (reflecting uncertainty in the tree inference or genuine discordance among the loci analysed). Existing tree visualisation tools are however not well suited to these tasks. In particular, side-by-side comparison of trees can prove challenging beyond a few dozen taxa
Dendroscope
Allows users to draw and compare rooted phylogenetic networks. Dendroscope is a program that includes characteristics allowing researchers to treat realistic phylogenetic trees. It also provides an interactive platform for researchers to explore the application of rooted phylogenetic networks to their phylogenetic trees and data. Moreover, this tool is available with a graphical user interface (GUI) or a command line interface (CLI).
Publications
Documentation
Institution(s)
Center for Bioinformatics (ZBIT), University of Tubingen, Germany; Department of Computer Science at the University of Auckland, New Zealand
TreeKO
Enables the comparison of tree topologies, even in the presence of duplication and loss events. To do so treeKO recursively splits gene trees into pruned trees containing only orthologs to subsequently compute a distance based on the combined analyses of all pruned tree comparisons. In addition, treeKO implements the possibility of computing phylome support values, and reconciliation-based measures such as the number of inferred duplication and loss events.
Publications
- (2011 Nucleic Acids Res) TreeKO: a duplication-aware algorithm for the comparison of phylogenetic trees
Institution(s)
Bioinformatics and Genomics Programme, Centre for Genomic Regulation (CRG), UPF, Barcelona, Spain