Table of Contents
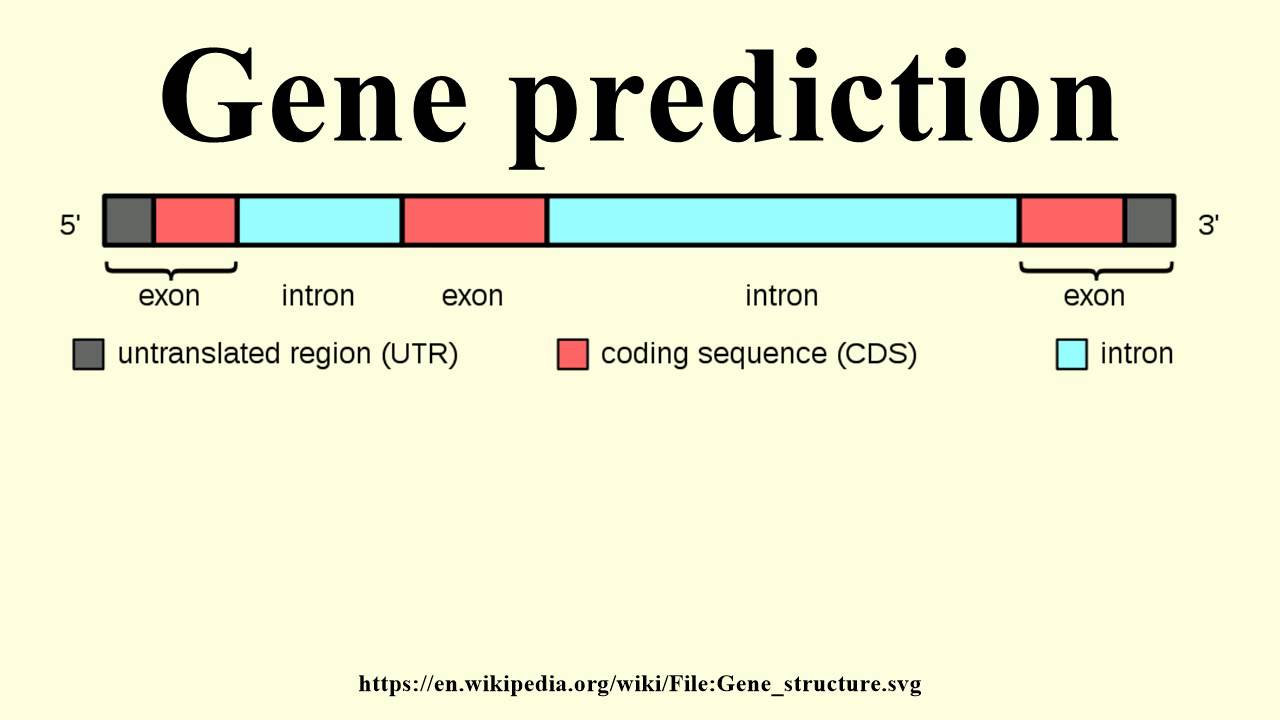
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.
Determining that a sequence is functional should be distinguished from determining the function of the gene or its product. Predicting the function of a gene and confirming that the gene prediction is accurate still demands in vivo experimentation[1] through gene knockout and other assays, although frontiers of bioinformatics research[citation needed] are making it increasingly possible to predict the function of a gene based on its sequence alone.
Gene prediction is one of the key steps in genome annotation, following sequence assembly, the filtering of non-coding regions and repeat masking.
The details about Empirical methods and Ab initio methods.
GeneMark
GeneMark developed in 1993 was the first gene finding method recognized as an efficient and accurate tool for genome projects. GeneMark was used for annotation of the first completely sequenced bacteria, Haemophilus influenzae, and the first completely sequenced archaea, Methanococcus jannaschii. The GeneMark algorithm uses species specific inhomogeneous Markov chain models of protein-coding DNA sequence as well as homogeneous Markov chain models of non- coding DNA. Parameters of the models are estimated from training sets of sequences of known type. The major step of the algorithm computes a posteriory probability of a sequence fragment to carry on a genetic code in one of six possible frames (including three frames in complementary DNA strand) or to be “non-coding”.
The GeneMark series could be used to:
- Gene Prediction in Bacteria, Archaea, Metagenomes and Metatranscriptomes
- Gene Prediction in Eukaryotes
- Gene Prediction in Transcripts
- Gene Prediction in Viruses, Phages and Plasmids
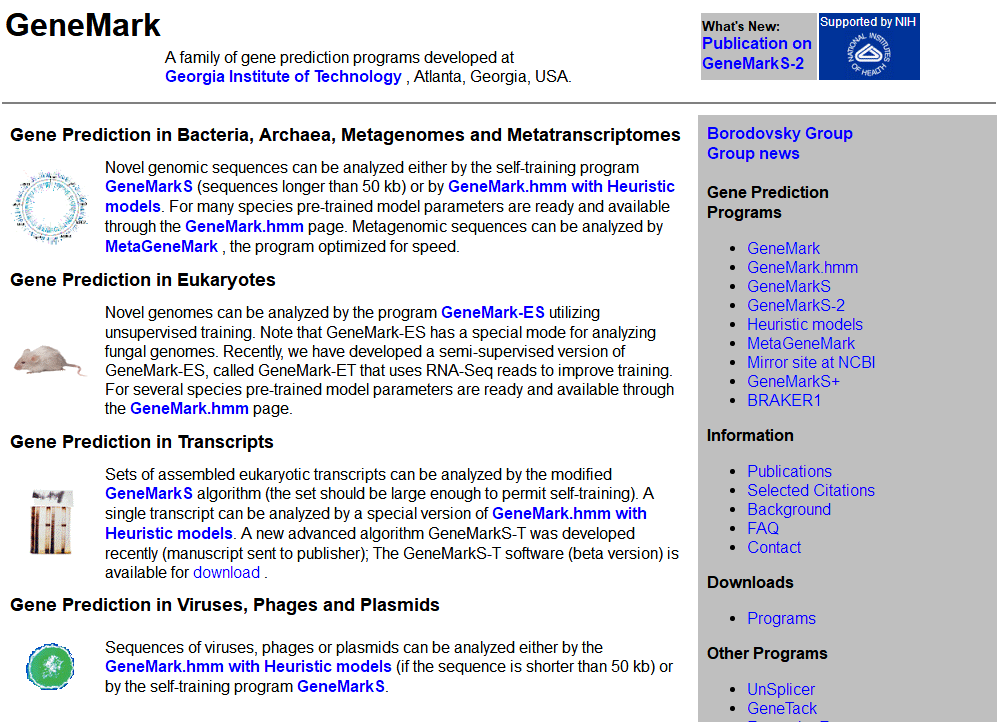
All the software programs are available for download and local installation.
GenScan
Identifies complete exon/intron structures of genes in genomic DNA. GENSCAN uses a homogeneous fifth order Markov model of noncoding regions and a three periodic (inhomogeneous) fifth order Markov model of coding regions. Features of the program include the capacity to predict multiple genes in a sequence, to deal with partial as well as complete genes, and to predict consistent sets of genes occurring on either or both DNA strands.
Official website: This server provides access to the program Genscan for predicting the locations and exon-intron structures of genes in genomic sequences from a variety of organisms. This server can accept sequences up to 1 million base pairs (1 Mbp) in length. If you have trouble with the web server or if you have a large number of sequences to process, request a local copy of the program (see instructions at the bottom of this page).
FGENESH
FGENESH - Program for predicting multiple genes in genomic DNA sequences.
FGENESH is the fastest (50-100 times faster than GenScan) and most accurate gene finder available.HMM-based gene structure prediction (multiple genes, both chains). Plant Molecular Biology (2005), 57, 3, 445-460: “Five ab initio programs (FGENESH, GeneMark.hmm, GENSCAN, GlimmerR and Grail) were evaluated for their accuracy in predicting maize genes. FGENESH yielded the most accurate and GeneMark.hmm the second most accurate predictions” (FGENESH identified 11% more correct gene models than GeneMark on a set of 1353 test genes).
Web version of FGENESH can be used with parameters for the following genomes: human, mouse, Drosophila, nematode, dicot plants, monocot plants, yeast (S.pombe) and Neurospora.
Reference: Solovyev V, Kosarev P, Seledsov I, Vorobyev D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006,7, Suppl 1: P. 10.1-10.12.
EMBOSS Cpgplot
EMBOSS Cpgplot identifies CpG islands in one or more nucleotide sequences. The ratio of observered to expected number of GC dinucleotides patterns is calculated over a window (sequence region) which is moved along the sequence. The calculated ratios are plotted graphically, together with the regions which match this program’s definition of a “CpG island” (a CG dinucleotide rich area). A report file is written giving the input sequence name, CpG island parameters and data on any CpG islands that are found.
Splign
Splign was developed with the following goals in mind:
- Accuracy in determining splice signals
- Recognition of short exons and non-consensus splices where feasible
- Ability to identify and separate multiple compartments typically representing gene copying events
- Production-grade performance
Internally at NCBI Splign is used to compute transcript alignments as a part of NCBI Genome Annotation Pipeline.
Splign comes in several flavors. If you are new to Splign, you can give it a try by navigating to Online page, typing in your accessions or GIs and submitting the job. For a more routine use, download and install the console version which is available for major platforms (and may also be available for a few platforms not listed - please request). You can also link to Splign from your own applications in a portable way since Splign is a part of the NCBI C++ Toolkit. And finally, Splign is available as a plugin for the NCBI Genome Workbench.
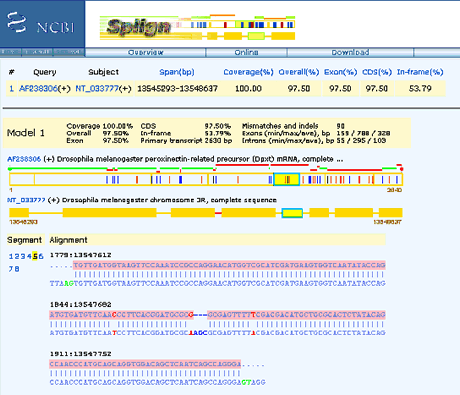
The online and algorithm could be found at Splign