Table of Contents
Protein Interactions Analysis
Proteins interact with nucleic acids, other proteins, ligands, metal ions, to exert their biological functions. Mass-spectrometry and protein chips have revolutionized the field of proteomics and the study of protein interactions to a wider scale, which require specific tools for their analyses. The study of protein interactions has implications for drug discovery and design.
Protein-DNA Interaction Prediction
Protein-DNA complexes play vital roles in many cellular processes by the interactions of amino acids with DNA. Several computational methods have been developed for predicting the interacting residues in DNA-binding proteins using sequence and/or structural information.
LigParGen | Protein-ligand docking : Protein interaction analysis
Provides an intuitive interface for generating OPLS-AA/1.14*CM1A(-LBCC) force field (FF) parameters for organic ligands. LigParGen is a web server which generates ligand parameters for common simulation software packages such as NAMD, GROMACS, OpenMM, BOSS and MCPRO. The software allows the users to obtain high quality parameters for molecular mechanics (MM) simulations without extensive knowledge about MM force fields or quantum mechanics (QM) methods.
Publications:
- (2017 Nucleic Acids Res) LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands.
Institutions(s):
Department of Chemistry, Yale University, New Haven, CT, USA
DBSI | DNA Binding Site Identifier
A powerful structure-based SVM model for the prediction and visualization of DNA binding sites on protein structures. DBSI is a machine learning approach to classify surface residues as binders or non-binders of DNA. DBSI employs sequence- and structure-based features encompassing a range of physical, chemical, geometric and evolutionary properties of the protein surface. DBSI also implements microenvironment features that allow for small-scale structural perturbation and the role of non-local cooperative effects. DBSI has been shown to be a top-performing model to predict DNA binding sites on the surface of a protein or peptide and shows promise in predicting RNA binding sites.
Publications:
- (2016 Bioinformatics) DBSI server: DNA binding site identifier.
Institutions(s):
Department of Biochemistry, University of Wisconsin-Madison, Madison, WI, USA; School of Life Sciences, Anhui University, Hefei, Anhui Province, China
Protein-RNA Interaction Prediction
Interactions between proteins and RNA play essential roles for life. For example, protein-RNA interactions mediate RNA metabolic processes such as splicing, polyadenylation, messenger RNA stability, localization and translation. Furthermore, many of these RNA-binding proteins are involved in human diseases.
HADDOCK | High Ambiguity Driven protein-protein DOCKing
An information-driven flexible docking approach for the modeling of biomolecular complexes. HADDOCK distinguishes itself from ab-initio docking methods in the fact that it encodes information from identified or predicted protein interfaces in ambiguous interaction restraints (AIRs) to drive the docking process. HADDOCK can deal with a large class of modeling problems including protein-protein, protein-nucleic acids and protein-ligand complexes.
Publications:
(2018 Nat Protoc) Defining distance restraints in HADDOCK.
(2016 J Mol Biol) The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes.
Institutions(s):
Bijvoet Center for Biomolecular Research, Science Faculty, Utrecht University, Utrecht, Netherlands
Global Score | Protein-RNA interactions
Predicts protein interactions with transcripts > 1000nt. Global Score is an algorithm that integrates the information coming from protein and RNA fragments into an overall binding propensity value. It was applied to all RBP-RNA pairs studied by eCLIP, and the number of predicted interactions significantly increases with the read counts, while pairs that are predicted to not interact show the opposite trend. It was also used to predict physical interactions between Xist and RNA-binding proteins and identified 5 interactions with Spen, Hnrnpk, Hrnnpu/Saf-A, Lbr and Ptbp1.
Publications:
- (2016 Nat Methods) Quantitative predictions of protein interactions with long noncoding RNAs.
Institutions(s):
Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Barcelona, Spain; Universitat Pompeu Fabra (UPF), Barcelona, Spain
Drug-target Interaction Prediction
Identification of drug-target interactions is an important process in drug discovery. Although high-throughput screening and other biological assays are becoming available, experimental methods for drug-target interaction identification remain to be extremely costly, time-consuming and challenging even nowadays. Therefore, various computational models have been developed to predict potential drug-target associations on a large scale.
Open Targets | Drug-target interaction prediction : Protein interaction analysis
A data integration and visualization platform that provides evidence about the association of known and potential drug targets with diseases. Open Targets platform is designed to support identification and prioritization of biological targets for follow-up. Each drug target is linked to a disease using integrated genome-wide data from a broad range of data sources. The platform provides either a target-centric workflow to identify diseases that may be associated with a specific target, or a disease-centric workflow to identify targets that may be associated with a specific disease. Users can easily transition between these target- and disease-centric workflows. Open Targets platform provides free data access through your web browser or through an API (Application Programming Interface).
Publications:
- (2017 Nucleic Acids Res) Open Targets: a platform for therapeutic target identification and validation.
Institutions(s):
Open Targets, Wellcome Genome Campus, Hinxton, Cambridge, UK; GSK, Medicines Research Center, Gunnels Wood Road, Stevenage, UK; Biogen, Cambridge, MA, USA
PiHelper | Drug-target interaction prediction : Protein interaction analysis
A drug- and antibody-target information aggregator and provider service.
Publications:
- (2013 Bioinformatics) PiHelper: an open source framework for drug-target and antibody-target data.
Institutions(s):
Computational Biology Center, Memorial Sloan-Kettering Cancer Center, NY, USA
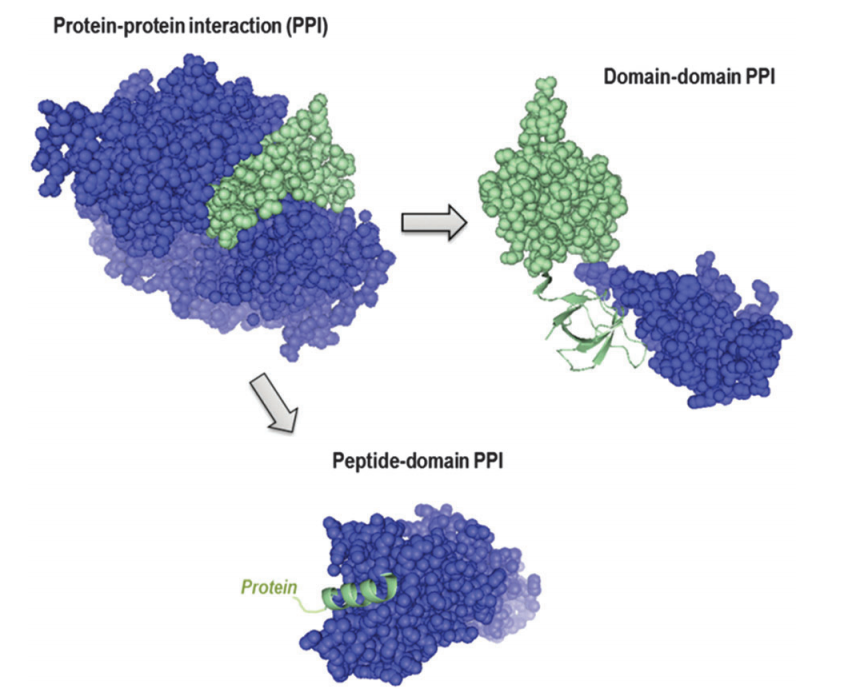
Protein-protein Interaction Prediction
Systems biology research is like solving a puzzle: the goal is to figure out how the various parts interact and work together. The interactome of an organism is then analogous to the puzzle’s key: it describes the network of all the protein–protein interactions (PPIs). As such, identifying all the PPIs for an organism is of great value. Despite the use of high-throughput techniques in discovering PPIs, however, the coverage of experimentally determined PPI data remains poor. Such low coverage is partly because the set of possible PPIs to be verified is so large that any exhaustive experimental verification will take a long time, even with high-throughput techniques.
mentha | Protein-protein interaction prediction : Protein interaction analysis
Provides protein-protein interaction (PPI) data for many species, including human. mentha allows users to assemble and analyze collections of proteins and networks of interest. It focuses on experimentally demonstrated physical interactions, in order to avoid the confusion between physical and genetic interactions and between experimental and inferred interactions. The database’s information come from manually curated protein-protein interaction databases.
Publications:
- (2013 Nat Methods) mentha: a resource for browsing integrated protein-interaction networks
Institutions(s):
Department of Biology, University of Rome Tor Vergata, Rome, Italy; Fondazione Santa Lucia Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
compPASS | comparative proteomic analysis software suite
Allows to analyze proteomic data. The compPASS method gives confidence measurements to interactions from parallel non-reciprocal proteomic datasets in using unbiased metrics. This tool is applicable to proteomic investigations ranging from focused studies on a small number of selected proteins to the analysis of entire protein families or biological regulatory networks.
Publications:
Institutions(s):
Department of Pathology, Harvard Medical School, Boston, MA, USA; Department of Cell Biology, Harvard Medical School, Boston, MA, USA
Protein-ligand Docking
Protein-ligand docking is a key computational method in the design of starting points for the drug discovery process.
UCSF Chimera
Permits to interactively visualize and analyse molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. UCSF Chimera allows users to incorporate new features. It contains some extensions which permits to visualize large-scale molecular assemblies such as viral coats, and allows researchers to share a Chimera session interactively despite being at separate locales. Other extensions can be used for extend the tool capabilities.
Publications:
- (2004 J Comput Chem) UCSF Chimera–a visualization system for exploratory research and analysis.
Institutions(s):
Computer Graphics Laboratory, Department of Pharmaceutical Chemistry, University of California, San Francisco, CA, USA
Protein-protein Docking
Protein-protein interactions lie at the heart of most cellular processes. Protein-protein docking is important for understanding disease mechanisms and for drug discovery.
PIPER | Protein-protein docking : Protein interaction analysis
Generates accurate structures of protein-protein complexes. PIPER is a Fast Fourier Transform (FFT)-based protein docking program, extended to be used with pairwise interaction potentials and based on docking code from the Vajda lab at Boston University. The software allows to improve results docking an antigen to an antibody or docking to form a dimer or trimer.
Publications:
Institutions(s):
Department of Biomedical Engineering, Boston University, Boston, MA, USA; Program in Bioinformatics, Boston University, Boston, MA, USA
HADDOCK | High Ambiguity Driven protein-protein DOCKing
An information-driven flexible docking approach for the modeling of biomolecular complexes. HADDOCK distinguishes itself from ab-initio docking methods in the fact that it encodes information from identified or predicted protein interfaces in ambiguous interaction restraints (AIRs) to drive the docking process. HADDOCK can deal with a large class of modeling problems including protein-protein, protein-nucleic acids and protein-ligand complexes.
Publications:
(2018 Nat Protoc) Defining distance restraints in HADDOCK.
(2010 Nat Protoc) The HADDOCK web server for data-driven biomolecular docking.
Institutions(s):
Bijvoet Center for Biomolecular Research, Science Faculty, Utrecht University, Utrecht, Netherlands