Table of Contents
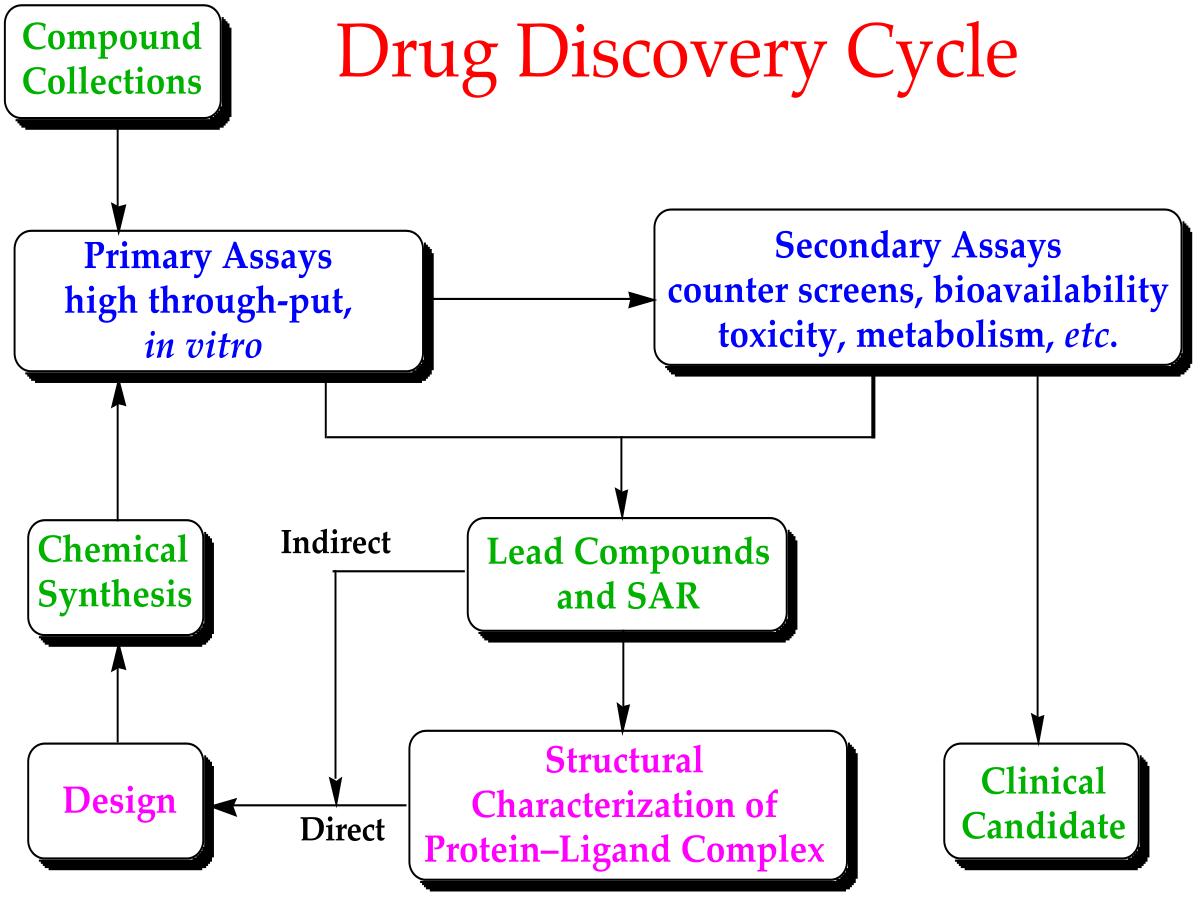
Drug Discovery
In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered. Historically, drugs were discovered through identifying the active ingredient from traditional remedies or by serendipitous discovery. Later chemical libraries of synthetic small molecules, natural products or extracts were screened in intact cells or whole organisms to identify substances that have a desirable therapeutic effect in a process known as classical pharmacology. Since sequencing of the human genome which allowed rapid cloning and synthesis of large quantities of purified proteins, it has become common practice to use high throughput screening of large compounds libraries against isolated biological targets which are hypothesized to be disease modifying in a process known as reverse pharmacology. Hits from these screens are then tested in cells and then in animals for efficacy.
The goal of drug discovery is to identify, optimize and clinically validate those compounds that bind and modulate the function of a target protein implicated in a disease state. A drug molecule must possess certain geometry and physicochemical properties in order to have a sufficiently high binding affinity toward a given macromolecular target. As a result, the number of bioactive compounds is very small compared to a vast collection of candidate compounds. Source
Drug-drug interaction prediction
Drug-drug interactions (DDIs) are a major cause of adverse drug effects and a public health concern, as they increase hospital care expenses and reduce patients’ quality of life. DDI detection is, therefore, an important objective in patient safety, one whose pursuit affects drug development and pharmacovigilance.
DDI-CPI | Drug-Drug Interaction via Chemical-Protein Interactome
A server Predicting Drug-Drug Interaction via Chemical-Protein Interactome.
Publications:
- (2014 Nucleic Acids Res) DDI-CPI, a server that predicts drug-drug interactions through implementing the chemical-protein interactome.
Institutions(s):
Bio-X Institutes, Shanghai Jiao Tong University, Shanghai, China;
University of Arkansas at Little Rock/University of Arkansas for Medical Sciences, Little Rock, AR, USA;
Healthcare Analytics Research Group, IBM TJ Watson Research Center, Yorktown Heights, NY, USA
Hierarchical RNNs model-based DDI extraction
Allows extraction of drug-drug interactions (DDI). Hierarchical RNNs model-based DDI extraction aims to identify and organize the drug-drug interaction in biomedical texts, which has been the state-of-the-art methods for DDI extraction task. It can effectively integrate the sequence and shortest dependency path (SDP) for DDI extraction.
Publications:
- (2017 Bioinformatics) Drug-drug Interaction Extraction via Hierarchical RNNs on Sequence and Shortest Dependency Paths.
Institutions(s):
College of Computer Science and Technology, Dalian University of Technology, Dalian, China;
Stanford Center for Biomedical Informatics Research, Stanford University, CA, USA
Ligand-based Virtual Screening
FTrees | Feature Trees
Handles and searches large virtual combinatorial libraries. FTrees performs similarity searches in the generated Fragment Space. It stores the physicochemical properties of the substructure represented by a node in a property profile for that node. This tool is able to select that topology-preserving mapping belonging to the best matching overlay of two molecules both represented by their corresponding Feature Trees.
Publications:
(2011 J Am Chem Soc) Software Review of FTrees and FTrees-FS in Pipeline Pilot.
(2009 J Chem Inf Model) Searching Fragment Spaces with feature trees.
Maintainer(s):
FTrees Team support@biosolveit.de
Frog | FRee On line druG conformation generation
Builds small 3D compounds. Frog can construct single or ensembles of low to medium energy 3D conformations starting from a 1D/2D or 3D given structure. It clarifies compound stereochemistry including chiral sites with a user-defined maximum number of generated conformers. This tool is useful for in silico studies, such as ligand-based or structure-based virtual screening; or lead compounds optimization.
Publications:
(2010 Nucleic Acids Res) Frog2: Efficient 3D conformation ensemble generator for small compounds.
(2007 Nucleic Acids Res) Frog: a FRee Online druG 3D conformation generator.
Institutions(s):
MTi, INSERM UMR-S973, Universite Paris Diderot - Paris 7, Paris, France; RPBS, Universite Paris Diderot - Paris 7, Paris, France
Drug Repositioning
Drug repositioning has been regarded as one of the most promising strategies for translational medicine. Common efforts to find new uses for existing drugs depend on text mining, chemical genetics and network analysis. In the April 2012 issue, Sanseau et al. proposed the use of genome-wide association studies (GWAS) for drug repositioning.
DRRS | Drug repositioning recommendation system
Identifies novel disease indications for given drugs. DRRS is a drug repositioning recommendation system approach that addresses the problem of drug repositioning, using low-rank matrix appropriation and randomized algorithm. The software can also be used for drugs without previously known disease associations and is able to handle large datasets.
Publications:
- (2018 Bioinformatics) Computational Drug Repositioning using Low-Rank Matrix Approximation and Randomized Algorithms.
Institutions(s):
School of Information Science and Engineering, Central South University, ChangSha, China;
Department of Computer Science, Old Dominion University, Norfolk, VA, USA
MANTRA | Mode of Action by NeTwoRk Analysis
Allows to analyze the Mode of Action (MoA) of novel drugs and to identify known and approved candidates for “drug repositioning”. MANTRA is a computational tool based on network theory and non-parametric statistics on gene expression data, which offers three “workspaces”: Analysis, Network and Search. Users can visually explore the Drug Network that provides, for each of the drugs, information about biochemical interactions, therapeutic indications, known MoA, pharmacology and targeted proteins.
Publications:
(2014 Bioinformatics) Mantra 2.0: an online collaborative resource for drug mode of action and repurposing by network analysis.
(2010 Proc Natl Acad Sci U S A) Discovery of drug mode of action and drug repositioning from transcriptional responses.
Institutions(s):
Telethon Foundation (TGM11SB1), the Italian Ministry of Health (GR-2009-1596824), the EU Project Nanosol (FP7/2007-2013 309329) and the Italian Ministry of Research (PON A3-00311).
Chemical Toxicity
Determining the toxicity of chemicals is necessary to identify their harmful effects on humans, animals, plants, or the environment. It is also one of the main steps in drug design.
ChemDes | Chemical toxicity : Drug design
An integrated web-based platform for molecular descriptor and fingerprint computation. ChemDes provides more than 3,679 molecular descriptors that are divided into 61 logical blocks. In addition, it provides 59 types of molecular fingerprint systems for drug molecules, including topological fingerprints, electro-topological state fingerprints, MACCS keys, FP4 keys, atom pairs fingerprints, topological torsion fingerprints and Morgan/circular fingerprints.
Publications:
- (2015 J Cheminform) ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation.
Institutions(s):
School of Pharmaceutical Sciences, Central South University, Changsha, China;
Department of Biostatistics, School of Public Health, University of Texas Health Science Center at Houston, Houston, USA
Molecular editors
A molecule editor, i.e. a program facilitating graphical input and interactive editing of molecules, is an indispensable part of every cheminformatics or molecular processing system.
Bioclipse | Molecule editors : Drug design
Provides 2D-editing, 3D-visualization, file format conversion, calculation of chemical properties, … Bioclipse delivers a stable, scalable integration platform for developers and an intuitive workbench for end users. The tool includes a rewritten editor for chemical structures, a table for multiple molecules that supports gigabyte-sized files, as well as a graphical editor for sequences and alignments. It provides advanced functionality in fields such as cheminformatics, bioinformatics, semantic web, spectrum analysis, drug discovery, safety assessment, and general chemistry education.
Publications:
(2009 BMC Bioinformatics) Bioclipse 2: a scriptable integration platform for the life sciences.
(2007 BMC Bioinformatics) Bioclipse: an open source workbench for chemo- and bioinformatics.
Institutions(s):
Department of Pharmaceutical Biosciences, Uppsala University, Uppsala, Sweden;
Wellcome Trust Genome Campus, European Bioinformatics Institute, Hinxton, UK;
Max von Pettenkofer-Institut, Ludwig-Maximilians-Universität, Munich, Germany
MCCE | Multi-Conformation Continuum Electrostatics
Combines continuum electrostatics (CE) and molecular mechanics force fields to calculate the equilibrium distribution of ionization states and atomic positions. MCCE is a biophysics simulation program that adds side chain and ligand conformational degrees of freedom to a CE analysis of pKas and Ems. The software allows side chain conformation sampling within pH titrations.
Publications:
(2009 J Comput Chem) MCCE2: improving protein pKa calculations with extensive side chain rotamer sampling.
(2002 Biophys J) Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins.
Institutions(s):
Department of Physics, City College of New York, New York, NY, USA