Table of Contents
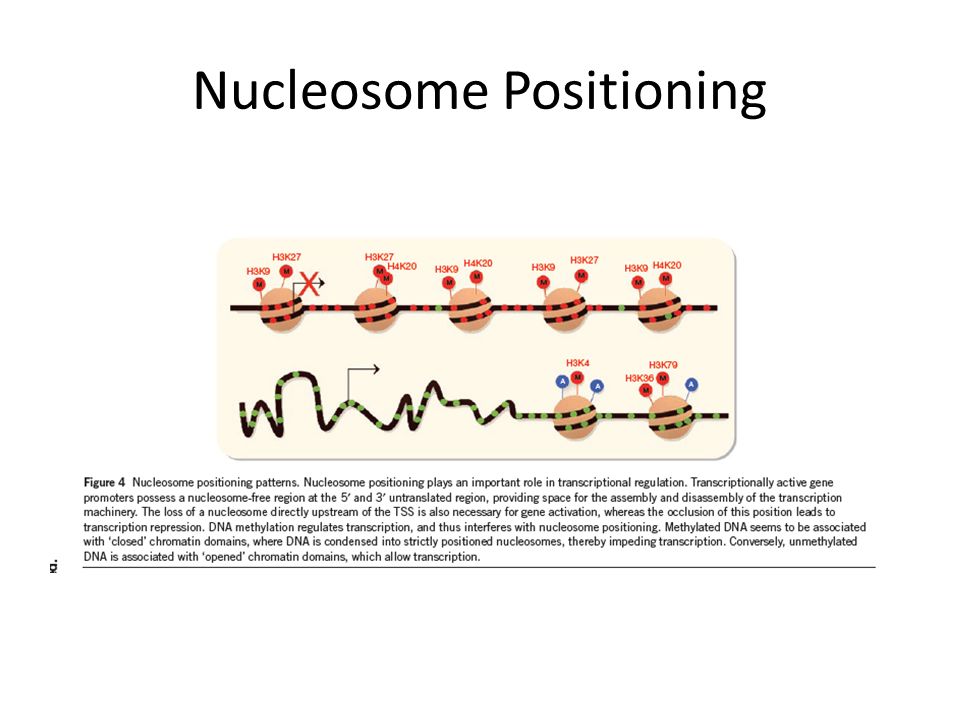
Nucleosome Positioning
NuCMap | Nucleosome positioning : Epigenomics
Maps directly nucleosome centers. NuCMap is based on chemical modification of engineered histones. It locates nucleosome positions genome-wide on a map in unprecedented detail and accuracy and shows significantly stronger dinucleotide signals in nucleosome DNAs than MNase maps. The tool reveals novel aspects of the in vivo nucleosome organization that are linked to transcription factor (TF) binding, RNA polymerase pausing, and the higher order structure of the chromatin fiber.
Publications:
- (Brogaard, 2012) A map of nucleosome positions in yeast at base-pair resolution. Nature.
Institutions(s):
Department of Molecular Biosciences and Department of Chemistry, Northwestern University, Evanston, IL, USA; Department of Statistics, Northwestern University, Evanston, IL, USA
nuMap | Nucleosome positioning : Epigenomics
Implements the YR and W/S schemes to predict nucleosome positioning at high resolution. This methodology is based on the sequence-dependent anisotropic bending, which dictates how DNA is wrapped around a histone octamer. nuMap allows users to specify a number of options such as schemes and parameters for threading calculation and provides multiple layout formats.
Publications:
- (Cui and Zhurkin, 2014) Rotational positioning of nucleosomes facilitates selective binding of p53 to response elements associated with cell cycle arrest. Nucleic Acids Res.
- (Cui and Zhurkin, 2010) Structure-based analysis of DNA sequence patterns guiding nucleosome positioning in vitro. J Biomol Struct Dyn.
Institutions(s):
Thomas H Gosnell School of Life Sciences, Chinahester Institute of Technology, Rochester, NY, USA
ICM | Interactive Chromatin Modeling
Allows users to assess nucleosome stability and fold sequences of DNA into putative chromatin templates. ICM generates a nucleosome energy level diagram, coarse-grained representations of free DNA and chromatin and plots of the helical parameters (Tilt, Roll, Twist, Shift, Slide and Rise) as a function of position. It uses an elastic model to automatically place nucleosomes. The tool can be used to y assemble models of chromatin that can be employed to rationalize biophysical data, especially spatial relations.
Publications:
- (Stolz and Bishop, 2010) ICM Web: the interactive chromatin modeling web server. Nucleic Acids Res.
Institutions(s):
Department of Biostatistics, Tulane University, New Orleans, LA, USA; Centre for Computational Science, Lindy Boggs Centre, Tulane University, New Orleans, LA, USA
MNase-seq analysis
The technology of micrococcal nuclease (MNase) digestion combined with high-throughput sequencing (MNase-seq) is a powerful method to map the genome-wide distribution of nucleosome occupancy (Mavrich et al., 2008; Schones et al., 2008; Jiang and Pugh, 2009). Source text: Chen et al., 2014.
PING
Serves for nucleosome positioning. PING uses MNase-Seq data or MNase - or sonicated - ChIP-Seq data combined with either single-end or paired-end sequencing. It enables nucleosome predictions even in the presence of low read counts. It contains functions for pre-processing and reading raw data. It can handle sonicated and MNase protocols combined with either single-end (SE) or paired-end (PE) sequencing.
Publications:
- (Woo et al., 2013) PING 2.0: an R/Bioconductor package for nucleosome positioning using next-generation sequencing data. Bioinformatics.
- (Zhang et al., 2012) Probabilistic inference for nucleosome positioning with MNase-based or sonicated short-read data. PLoS One.
Institutions(s):
Vaccine and Infectious Diseases and Public Health Sciences Divisions, Fred Hutchinson Cancer Research Center, Seattle, WA, USA; Department of Biostatistics, Johns Hopkins University, Baltimore, MD, USA;
nucleR
A package for non-parametric nucleosome positioning. nucleR uses a novel aproach in this field which comprises a deep profile cleaning using Fourier Transform and peak scoring for a quick and flexible nucleosome calling.
Publications:
- (Flores and Orozco, 2011) nucleR: a package for non-parametric nucleosome positioning. Bioinformatics.
Institutions(s):
IRB-BSC Joint Research Program on Computational Biology, Institute of Research in Biomedicine, Barcelona, Spain
NOrMAL
A command line tool for accurate placing of the nucleosomes. NOrMAL was designed to resolve overlapping nucleosomes and extract extra information (“fuzziness”, probability, etc.) of nucleosome placement.
Publications:
- (Polishko et al., 2012) NORMAL: accurate nucleosome positioning using a modified Gaussian mixture model. Bioinformatics.
Institutions(s):
Department of Computer Science and Engineering, University of California, Riverside, CA, USA
ATAC-seq analysis
ATAC-seq allows simultaneous interrogation of factor occupancy, nucleosome positions in regulatory sites, and chromatin accessibility genome wide by considering the position of insertion and the distribution of insert lengths captured during the transposition reaction.
NucleoATAC | Nucleosome positionning analysis : ATAC-seq analysis
A python package for nucleosome-positioning using as basis the highly structured pattern of DNA fragment lengths and positions around nucleosomes. NucleoATAC can identify the rotational and translational positions of nucleosomes with up to base-pair resolution and provide quantitative measures of nucleosome occupancy in Schizosaccharomyces cerevisiae, Schizosaccharomyces pombe and human cells. NucleoATAC can be used to analyze sequence features underlying nucleosome positioning, to promote chromatin architecture across species, to identify transient changes in nucleosome occupancy and to asses positioning during a dynamic cellular response.
Publications:
- (Schep et al., 2015) Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res.
Institutions(s):
Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA; Biophysics Program, Stanford University School of Medicine, Stanford, CA, USA; Department of Applied Physics, Stanford University, Stanford, CA, USA